

|

On the Road to Treating Mitochondrial Disease (December 2021)

|
Recent advancements in mitochondrial genome editing technologies take scientists one step closer to developing viable treatments for mitochondrial diseases, which affect 1 in 4300 adults.
DRUGDISCOVERYNEWS.COM |
New Tool to Manipulate mtDNA in vivo (May 2021)

Precision BioSciences Announces New Study Published in Nature Communications Using Engineered ARCUS Nuclease to Target Mutant Mitochondrial DNA In Vivo.
DURHAM, N.C.--(BUSINESS WIRE)--Jun. 1, 2021-- Precision BioSciences, Inc. (Nasdaq: DTIL), a clinical stage biotechnology company developing allogeneic CAR T and in vivo gene correction therapies with its ARCUS® genome editing platform, today announced a new paper published online in Nature Communications that reports preclinical results using an ARCUS nuclease to target mitochondrial DNA (mtDNA) and reduce levels of mutant mtDNA in vivo.
The study, “Mitochondrial targeted meganuclease as a platform to eliminate mutant mtDNA in vivo” was led by Carlos T. Moraes, Ph.D., Esther Lichtenstein Professor in Neurology at the University of Miami Miller School of Medicine, with Ugne Zekonyte as first author.
Mitochondrial disorders impair the function of mitochondria, the organelles that produce the energy needed by cells. Organs and tissues that require more energy, such as the heart, muscles and brain, are more often affected. Additionally, both mutant and wild-type mtDNA can co-exist within the mitochondria of a cell, a phenomenon called mtDNA heteroplasmy. When specific threshold levels of mutant mtDNA are reached, cell function can be compromised, and disease can manifest1. It is believed that a shift in mtDNA heteroplasmy toward wild-type may provide therapeutic benefit for patients, and not all mutant mtDNA must be eliminated to achieve improvements in symptoms; mutant mtDNA levels just need to be reduced below the disease threshold level.
“In the past, mitochondrial-targeted nucleases have been successful in shifting mtDNA heteroplasmy but have come with unwanted drawbacks, most notably large size, heterodimeric nature, inability to distinguish single base changes, or low flexibility and effectiveness,” said Dr. Moraes. “In this study, a mitochondrial-targeted ARCUS nuclease (mitoARCUS) used to edit mutant mtDNA was particularly effective, in part because of the nuclease’s small size and single protein nature. We are very excited with this early research and the great promise we believe it suggests for using ARCUS editing in patients with mtDNA diseases in the future.”
Researchers involved with the study reported mitoARCUS-induced heteroplasmic shifts of up to 60% in vitro, with changes persisting for up to three weeks. When tested in a heteroplasmic mouse model, mitoARCUS delivered by AAV effectively shifted heteroplasmy towards wild-type in several of the analyzed tissues of juvenile mice, with no depletion in total mtDNA levels at 6, 12, or 24 weeks. In adult mice treated with AAV-mitoARCUS, there was no editing at any of the potential nuclear off-target sites, and liver and skeletal muscle showed robust elimination of mutant mtDNA with concomitant restoration of mitochondrial transfer RNA levels.
“This is the first time ARCUS has been used to edit outside the nuclear genome and has done so with encouraging safety and efficacy in this mouse model,” said Derek Jantz, Ph.D., co-author of the paper and Chief Scientific Officer at Precision BioSciences. “We continue to see promising results in preclinical studies suggesting that ARCUS could potentially effectively edit mutant mtDNA in vivo in human clinical trials. I congratulate Carlos and his team on this research and look forward to further work on this program.”
Dr. Moraes will discuss this paper during the United Mitochondrial Disease Foundation’s Mitochondrial Medicine 2021 Virtual “Meet the Scientific Program Faculty” on Friday, June 4, 2021 at 12:00 PM EDT.
The study, “Mitochondrial targeted meganuclease as a platform to eliminate mutant mtDNA in vivo” was led by Carlos T. Moraes, Ph.D., Esther Lichtenstein Professor in Neurology at the University of Miami Miller School of Medicine, with Ugne Zekonyte as first author.
Mitochondrial disorders impair the function of mitochondria, the organelles that produce the energy needed by cells. Organs and tissues that require more energy, such as the heart, muscles and brain, are more often affected. Additionally, both mutant and wild-type mtDNA can co-exist within the mitochondria of a cell, a phenomenon called mtDNA heteroplasmy. When specific threshold levels of mutant mtDNA are reached, cell function can be compromised, and disease can manifest1. It is believed that a shift in mtDNA heteroplasmy toward wild-type may provide therapeutic benefit for patients, and not all mutant mtDNA must be eliminated to achieve improvements in symptoms; mutant mtDNA levels just need to be reduced below the disease threshold level.
“In the past, mitochondrial-targeted nucleases have been successful in shifting mtDNA heteroplasmy but have come with unwanted drawbacks, most notably large size, heterodimeric nature, inability to distinguish single base changes, or low flexibility and effectiveness,” said Dr. Moraes. “In this study, a mitochondrial-targeted ARCUS nuclease (mitoARCUS) used to edit mutant mtDNA was particularly effective, in part because of the nuclease’s small size and single protein nature. We are very excited with this early research and the great promise we believe it suggests for using ARCUS editing in patients with mtDNA diseases in the future.”
Researchers involved with the study reported mitoARCUS-induced heteroplasmic shifts of up to 60% in vitro, with changes persisting for up to three weeks. When tested in a heteroplasmic mouse model, mitoARCUS delivered by AAV effectively shifted heteroplasmy towards wild-type in several of the analyzed tissues of juvenile mice, with no depletion in total mtDNA levels at 6, 12, or 24 weeks. In adult mice treated with AAV-mitoARCUS, there was no editing at any of the potential nuclear off-target sites, and liver and skeletal muscle showed robust elimination of mutant mtDNA with concomitant restoration of mitochondrial transfer RNA levels.
“This is the first time ARCUS has been used to edit outside the nuclear genome and has done so with encouraging safety and efficacy in this mouse model,” said Derek Jantz, Ph.D., co-author of the paper and Chief Scientific Officer at Precision BioSciences. “We continue to see promising results in preclinical studies suggesting that ARCUS could potentially effectively edit mutant mtDNA in vivo in human clinical trials. I congratulate Carlos and his team on this research and look forward to further work on this program.”
Dr. Moraes will discuss this paper during the United Mitochondrial Disease Foundation’s Mitochondrial Medicine 2021 Virtual “Meet the Scientific Program Faculty” on Friday, June 4, 2021 at 12:00 PM EDT.
Gene Therapy Can Now Treat Mitochondrial Diseases
Illness-causing mutations in mitochondrial DNA (mtDNA) happen in around 1 of every 5,000 adults and result in serious and often fatal conditions ranging from muscle weakness to coronary diseases. Even though three-parent IVF—a dubious technique that uses a third individual’s mitochondria notwithstanding a mother’s egg nucleus and a father’s sperm—has been proposed as an approach to avoid the legacy of mtDNA changes, there are at present no medications for a person born with the deformities.
CRISPR gene-editing approach has achieved extraordinary headway in efforts to alter changes found in nuclear DNA, yet scientists have experienced issues applying a similar approach to mtDNA in light of the fact that mitochondria appear not to take up the guide RNAs that enable the tool to home in on the correct sequence. In the two most recent studies, scientists rather used older gene-editing approaches that don’t require guide RNAs.
In one study, the University of Miami’s Carlos Moraes and colleagues infused adeno-related infections containing mitochondrial-targeted on TALENs into the muscles of mice carrying a mtDNA change. Following a half year, levels of mutant mtDNA in the creatures’ muscle tissue had dropped by more than 50 percent—below the levels generally associated with indications of the mitochondrial disease.
CRISPR gene-editing approach has achieved extraordinary headway in efforts to alter changes found in nuclear DNA, yet scientists have experienced issues applying a similar approach to mtDNA in light of the fact that mitochondria appear not to take up the guide RNAs that enable the tool to home in on the correct sequence. In the two most recent studies, scientists rather used older gene-editing approaches that don’t require guide RNAs.
In one study, the University of Miami’s Carlos Moraes and colleagues infused adeno-related infections containing mitochondrial-targeted on TALENs into the muscles of mice carrying a mtDNA change. Following a half year, levels of mutant mtDNA in the creatures’ muscle tissue had dropped by more than 50 percent—below the levels generally associated with indications of the mitochondrial disease.
Advances in Gene Editing and Delivery Offer Promise for People Living with Mitochondrial Diseases

ADDITIONAL MEDIA NOTES
|
There is strength in numbers — or at least the right proportion. A new study from University of Miami Miller School of Medicine researchers demonstrates an ability to target and reduce debilitating high levels of mutant mitochondrial DNAin heart and other muscle tissue to an extent — so far in mice — that could be curative.
The findings offer hope to people living with relatively rare mitochondrial diseases, such as Kearns-Sayre syndrome, progressive external ophthalmoplegia, myoclonus epilepsy with ragged-red fibers, and Pearson’s syndrome. The DNA inside mitochondria — the “power plants” that generate energy in most cells and allow proper functioning — is present in multiple copies, and mutations cause diseases only when present in the vast majority of these molecules. People with high levels of mutated mitochondrial DNA (mtDNA) can experience muscle weakness, developmental delays, seizures, and other serious adverse effects. For years, researchers have searched for an effective way to reduce the high number of mutant mtDNA molecules in critical organs, like the heart, and in skeletal muscle. In a new study published in Nature Medicine, Carlos T. Moraes, Ph.D., professor of neurology and cell biology, and the Esther Lichtenstein Chair in Neurology, lead author Sandra R. Bacman, Ph.D., associate scientist in the Department of Neurology, and their colleagues offer a promising way to accomplish precisely that. READ MORE. |
Science Latest News
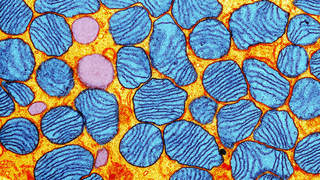
Zapping mutant DNA in mitochondria could treat major class of genetic disease.
By Mitch Leslie. Sep. 24, 2018
|
CRISPR, the genome editor celebrated as a potentially revolutionary medical tool, isn’t omnipotent. Mitochondria, the organelles that supply a cell’s energy, harbor their own mitochondrial DNA (mtDNA) and mutations there can have devastating consequences including deafness, seizures, and muscle weakness. Genome editing might be a remedy, but mitochondria appear to be off-limits to CRISPR.
Now, two studies published this week in Nature Medicine reveal that two older genome-editing tools can slash the amount of defective mtDNA in mice bred to have a mitochondrial disease, counteracting the effects of the mutation. The proof-of-principle results could open the way for the first treatments for mitochondrial diseases. “These are remarkable findings that make it possible to even consider doing this in humans,” says mitochondrial biologist Martin Picard of the Columbia University Irving Medical Center, who was not involved in the work. READ MORE |
RESEARCH HIGHLIGHT
ATAD3 helps shaping cristae of mitochondria
J Cell Sci 2018 131: e1301
|
The inner mitochondrial membrane ATPase family AAA domain-containing protein 3 (ATAD3) has several cellular roles, as shown in numerous organisms. In their Research Article (Peralta et al., 2018), Carlos Moraes and colleagues use a mouse strain with a conditional knockout that is specific to skeletal muscles to address the in vivo role of ATAD3. They find that loss of ATAD3 in muscles leads to progressive myopathy after 2-3 months in these mice. Furthermore, the cristae structure of mitochondria is disrupted and the surface area of cristae decreased. In absence of ATAD3, protein levels of the cristae biogenesis complex mitochondrial contact site and cristae organising system (MICOS) are reduced. The authors show that this, in turn, leads to changes in cholesterol metabolism, depletion of mitochondrial DNA (mtDNA) and impaired mtDNA replication. Interestingly, ATAD3 is not crucial for mitochondrial translation, since the observed decrease of levels in oxidative phosphorylation (OXPHOS) complex members occurs after the defect in cristae appearance. This work establishes the in vivo function of ATAD3 as mediating the ultrastructure of mitochondria and the control of key regulators of cristae morphology, such as MICOS, and, subsequently, the mtDNA replication process and metabolism.
|
MDA grant awarded to the Moraes Lab (Summer 2018)
|
|
Mitochondrial Myopathies – Carlos T. Moraes, PhD
“There are no treatments for mitochondrial diseases. The unique genetics of mitochondria offer some opportunities for genetic manipulation that could be curative. Our approach has the potential to be curative. The main limitation will be delivery systems for the DNA editing enzyme, which are being developed at a fast pace.” Carlos T. Moraes, professor of Neurology at the University of Miami’s Miller School of Medicine in Florida, was awarded an MDA Research Grant totaling $300,000 over 3 years to study monomeric gene-editing enzymes to treat mitochondrial myopathies. Mitochondrial diseases caused by mutations in the mitochondrial DNA (mtDNA) are most often heteroplasmic, meaning that the normal mtDNA coexists with the mutant mtDNA. Disease is manifested when the cells/tissue have a very high percentage of mutant mtDNA. There are no treatments for mitochondrial diseases, but the unique genetics of mitochondria offer some opportunities for genetic manipulation that could be curative. With previous MDA funding, Dr. Moraes and colleagues have developed approaches to reduce the percentage of mutant mtDNA using DNA-editing enzymes, but the feasibility of this approach is limited because these enzymes are difficult to deliver to affected tissues. Therefore, he has developed an innovative approach to improve the delivery of gene-editing components to mitochondria, and in this project will test the success in different animal models, improving the chances of bringing this approach to the clinics. |
Carlos T. Moraes Honored with Esther Lichtenstein Chair in Neurology
|
Carlos T. Moraes, Ph.D., professor of neurology and cell biology at the University of Miami Miller School of Medicine, was honored with the presentation of the Esther Lichtenstein Chair in Neurology. In his 20 years at the Miller School, Moraes has made significant advancements in understanding the cellular mechanisms behind degenerative neurological conditions, such as Parkinson’s and Alzheimer’s disease. Read more here.
|

Carlos T. Moraes, Ph.D., receives the Esther Lichtenstein Chair in Neurology, with, from left, Pascal J. Goldschmidt, M.D., Walter G. Bradley, D.M., F.R.C.P., Carlos T. Moraes, Ph.D. and Ralph L. Sacco. M.D., M.S.
|
