Manipulating mtDNA heteroplasmy
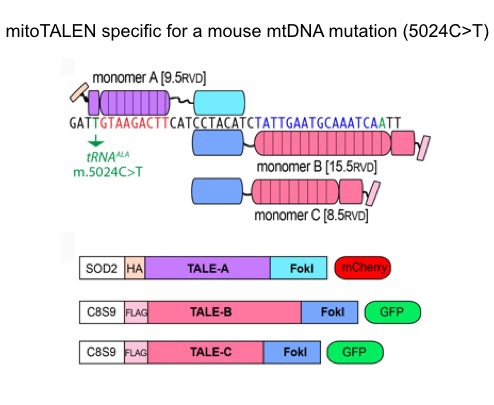
When mutant mtDNA is heteroplastic (i.e. co-exists with wild-type mtDNA), the ability to destroy the mutant genomes can increase the levels of wild-type mtDNA. We have developed a series of mtDNA editing enzymes designed to promote a double-strand break in the mtDNA. Once mtDNA is cleaved, it is rapidly degraded, leading to a transient depletion of mtDNA levels. The residual molecules (now with increased relative levels of wild-type mtDNA) repopulate mitochondria to normalize mtDNA levels. Recently, we have used mitoTALENs as tools to reduce levels of mutant mtDNA in defective cells and tissues (Pereira et al., 2018; Bacman et al., 2018).
mtDNA mutations in aging
As we age, small levels of mtDNA deletions accumulate in our cells, particularly in neurons and muscle. We have developed mouse models in which we can induce mtDNA damage in different tissues. These mice have been instrumental to analyze the role of mtDNA damage in the aging of skeletal muscle and neurons. In addition, we have studied how transcriptional regulators can compensate for age-related changes in gene expression (Garcia et al., 2018).
Understanding genotype–phenotype correlations in mitochondrial diseases
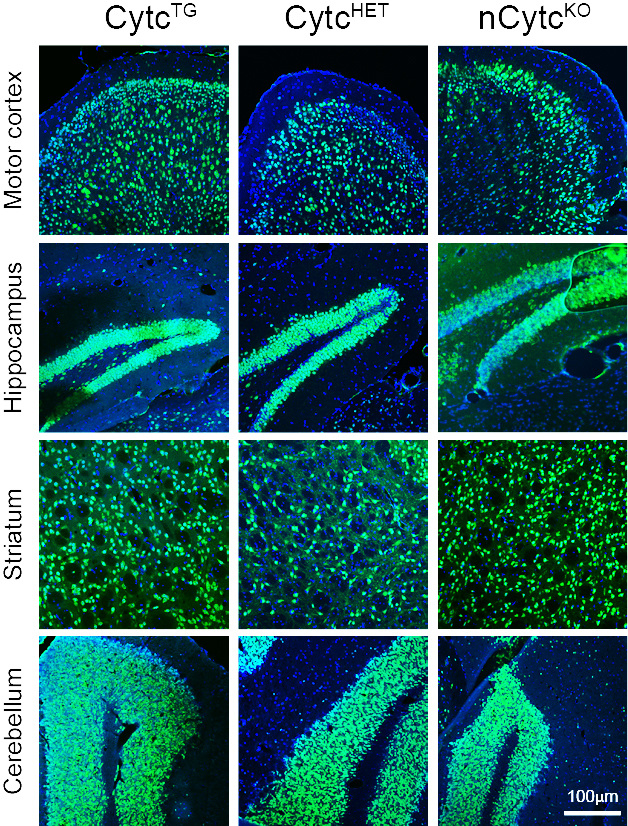
Mitochondrial diseases have heterogeneous clinical presentations. The causes of these phenotypes are poorly understood, and we have developed several models with defects in different mitochondrial oxidative phosphorylation enzymes to address this question, particularly for defects in the Central Nervous System. One such model is a mouse with defective cytochrome c in neurons, which showed a distinct phenotype (Pinto et al., 2018).
Molecular function of mitochondrial proteins
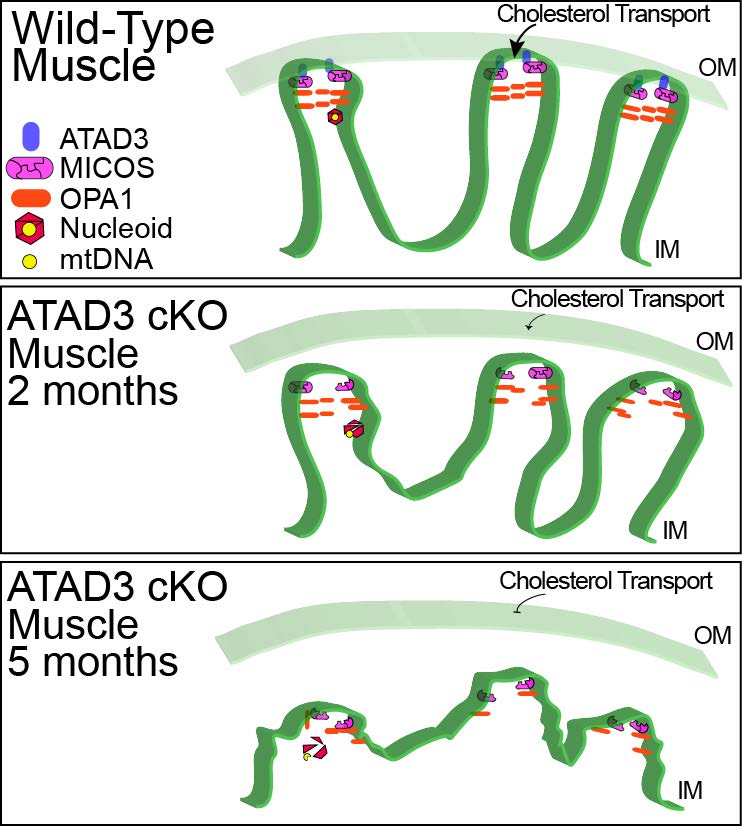
We also study the basic function of mitochondrial proteins that modulate the energy-producing oxidative phosphorylation system (Arguello et al., 2018). These include subunits of the OXPHOS complexes as well as proteins that indirectly affect mitochondrial function. We recently found that ATAD3 controls mitochondrial cristae structure and that loss of this protein destabilizes mitochondrial nucleoids, leading to mtDNA damage (Peralta et al., 2018).